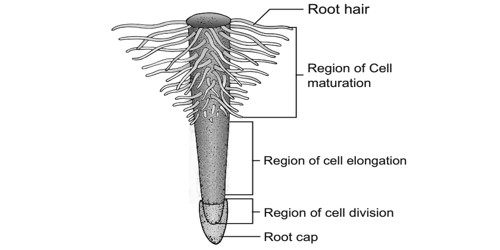
The root is covered at the apex by a thimble-like structure called the root cap (Figure). It protects the tender apex of the root as it makes its way through the soil. A few millimeters above the root cap is the region of meristematic activity. The cells of this region are very small, thin-walled and dense protoplasm. They divide repeatedly. The cells proximal to this region undergo rapid elongation and enlargement and are responsible for the growth of the root in length.
This region is called the region of elongation. The cells of the elongation zone gradually differentiate and mature. Hence, this zone, proximal to the region of elongation, is called the region of maturation. From this region some of the epidermal cells form very fine and delicate, thread-like structures called root hairs. These root hairs absorb water and minerals from the soil.
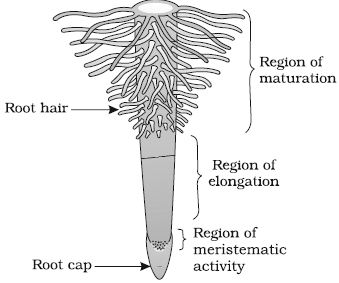
(i) Root cap region
It is a thimble-like formation produced by the meristematic zone and protects the tender apex from harsh soil particles. It protects the tender apex of the root as it makes its way through the soil. As the root extends further in the soil, the root cap wears out and renews continuously. It covers the apex of roots that grow through the soil.
(ii) Region of meristematic cells
It is a small region of actively dividing cells called the apical meristem. It consists of: Dermatogen, Periblem, and Plerome. These cells divide actively. This region is a few millimeters above the root cap. The cells in this region are thin-walled with dense protoplasm.
(iii) Region of elongation
This is placed next to the meristematic region, in which, the cells elongate and expand to make the root produce in length. This zone is accountable for the development in length of the root. Here the cells enlarge to make the root grow in length. The length is about 4-8 mm. These cells slowly differentiate and grown-up.
(iv) Region of maturation
This is subsequently to the area of elongation, in which, the cells grown-up and set apart into diverse tissues constituting. The zone of maturation is proximal to the zone of elongation. Root hair to absorb water and nutrients from the soil. It also gives rise to lateral roots.












